











一种TIM-3纳米抗体、制备方法及其应用
商品类型
专利
专利号
CN111253489A
所属行业
新材料技术
归属方
石河子大学
上架日期
2020-07-14 23:17:49
交易方式
转让
交易价格
面议
详细介绍
[0001] 本发明属于来自动物或人的抵抗物质技术领域,尤其涉及一种TIM-3纳米抗体、制备方法及其应用。
背景技术
[0002] 目前,T细胞免疫球蛋白黏蛋白分子-3(T-cell immunoglobulinandmucin-3,TIM-3),是一种膜蛋白,属于T细胞免疫球蛋白家族的新成员。首次发现是在Th1型淋巴细胞和CD8+T淋巴细胞中。鼠源的TIM-3编码基因存在于第11号染色体上,与人源TIM-3有很高的相似度,而人源的TIM-3编码基因存在于第5号染色体上,含有7个外显子。
[0003] 研究认为TIM-3只选择性的表达于激活的Th1细胞上,可以作为区分和Th2细胞的新的标志物。TIM-3通过与其配体结合能够发挥抑制Th1细胞的功能。它参与炎症的发生,能够调节免疫细胞的活化与功能,在许多疾病反应中起着非常重要的作用。在非Th1细胞上也会表达,并发挥着不同的生物学功能。当TIM-3与Bat3结合后,它的活性会被抑制。Th1特异性转录因子T-bet能调控TIM-3的表达,而且T-bet可以直接与TIM-3的启动子结合。TIM-3能够调节Th1与Th2细胞的功能,多种疾病的发生也与它的异常表达相关,因此被认为是机体疾病相关的重要基因。
[0004] 生物免疫治疗相对传统化疗或靶向治疗,有一个本质逻辑区别:“免疫治疗”针对的是免疫细胞(或者说是免疫系统),而不是癌症细胞。在癌症免疫治疗中,抑制免疫检查点通路被认为是最有前景的治疗方式之一,其机制是通过抑制通路中相关靶点解除T细胞活性受抑状态,活化后的T细胞能够进攻和消灭肿瘤细胞。抗体并不直接作用于肿瘤细胞,而是通过作用于T细胞类间接杀伤肿瘤细胞;另外,它们并不是针对肿瘤表面的某种特定物质,而是系统性地增强了全身的抗肿瘤免疫反应。TIM-3靶点以及TIM-3单克隆抗体是生物免疫治疗中的一个热点。
[0005] 纳米抗体是由比利时科学家于1993年在自然杂志中首次报道,在羊驼外周血液中存在一种天然缺失轻链的抗体,该抗体只包含一个重链可变区(VHH)和两个常规的CH2与CH3区,但却不像人工改造的单链抗体片段(scFv)那样容易相互沾粘,甚至聚集成块。更重要的是单独克隆并表达出来的VHH结构具有与原重链抗体相当的结构稳定性以及与抗原的结合活性,是目前已知的可结合目标抗原的最小单位。VHH晶体为2.5nm,长4nm,分子量只有15KD,因此也被称作纳米抗体(Nanobody,Nb)。
[0006] 综上所述,现有技术存在的问题是:
[0007](1)现有的单克隆抗体由于分子质量大150ku以及鼠源等问题对人体存在异源性。小鼠的单克隆抗体,对人体的应用会产生抗鼠单克隆抗体,它不可重复应用,影响其疗效。
[0008](2)现有的单克隆抗体体积相对较大,不能较好的进入肿瘤组织;单克隆抗体开发周期长,生产成本高,产量低;单克隆抗体类药物价格昂贵,研发复杂,人源化复杂,成功率有限。
[0009](3)现有的单克隆抗体难以大规模生产,单克隆抗体药物在建厂和生产过程中消耗的资金非常巨大。
[0010](4)现有的单克隆抗体不稳定,容易降解,保存成本高;容易污染,维护成本高昂。在高温和强酸强碱的条件下会分解,须在低温下保存,否则就会在数周内完全失去活性。抗体能被消化系统很快降解,从而阻止了其进入大脑或其他有效作用部位。
[0011] 解决上述技术问题的难度:纳米抗体包含3个高变区与4个骨架区,高变区均在同一侧。与人类抗体的VH有着同样的结构,测序显示与VH3同源性极高,但是纳米抗体的CDR1(Complementarity-determining region-1)与CDR3 (Complementarity-determining region-3)相对较长。纳米抗体的CDR3具有突出,可以增加与抗原结合的亲和力。纳米抗体具有稳定的结构,保证了结合的稳定性。噬菌体展示技术是应用普遍的一种生产纳米抗体的途径,将纳米抗体的序列导入到噬菌体序列中,目的蛋白将表达在噬菌体的外壳上,噬菌体文库构建是免疫驼科动物,取动物白细胞,将RNA进行反转录,构建对抗原有针对性地文库。
[0012] 解决上述技术问题的意义:中国医学科学院研制成一种具有定向性、选择性杀伤肿瘤细胞的“单抗-药物”联结物,即以单克隆抗体为“载体”,携带药物精确地和肿瘤细胞结合,能在原位杀死癌细胞,而对其他正常细胞无损伤。这种综合性药物好比是专攻癌症的“生物导弹”,人们把这种治疗方法形象地比喻为导弹疗法。而纳米抗体可以更好的进行这一功能。
[0013] 针对现有技术存在的问题,本发明提供了一种TIM-3抗原、纳米抗体及其筛选和鉴定方法、纳米抗体的应用。纳米抗体可以在严苛的环境中保持构象不变。纳米抗体抗热特别好,可以在室温下存放一周以上。而超强的耐酸碱性可以使纳米抗体更好的抵抗不同的环境,也可以增加纳米抗体的作用范围。单域重链抗体微小的体积也使它获得了较低的免疫原性。使动物长期注射蛋白成为可能。纳米抗体与抗原的结合位点也与单克隆抗体不同,并且可以结合到传统抗体结合不到的地方。纳米抗体可以使用原核表达生产,大大的降低了生产成本。
[0014] 本发明是这样实现的,一种纳米抗体,所述纳米抗体为TIM-3纳米抗体,其序列为SEQ ID NO:1。
[0015] 本发明的另一目的在于提供一种表达所述纳米抗体的TIM-3抗原,所述TIM-3抗原为哺乳动物细胞瞬时转染表达,使用TIM-3抗原对纳米抗体库进行多次筛选,获得特异性的纳米抗体噬菌体,测序得到目的片段。
[0016] 本发明的另一目的在于提供一种所述纳米抗体的筛选和鉴定方法,所述纳米抗体的筛选和鉴定方法包括以下步骤:
[0017] 第一步,构建载体:目的片段扩增,换载酶切,目的片段与载体连接,转化,筛选克隆。
[0018] 第二步,蛋白鉴定。
[0019] 第三步,构建抗体库:样品总RNA提取及cDNA合成,VHH文库片段的制备,电转化和文库构建。
[0020] 进一步,所述第一步的目的片段扩增具体为:
[0021](1)设计合成引物2条,通过PCR的方法扩增出足够量的目的产物。引物的序列为:SEQ ID NO:2、SEQ ID NO:3。
[0022](2)PCR反应采用的是pfu高温聚合酶。
[0023] 进一步,所述第一步的PCR各个成分的用量:引物浓度为1OD溶于400ul ddH2O。反应体系:50ul。引物mix,(1/28) 0.4ul x 13.6共8ul。10X pfu Buffer:5ul。每段上下游引物各2ul。Pfu:0.4ul(5u/ul)。ddH2O分别补水至50ul。
[0024] 目的片段PCR的方法扩增具体步骤为:
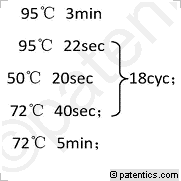
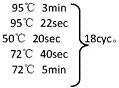
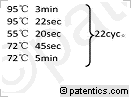
[0025](1)第一轮PCR程序:
[0026] /CN111253489A/CN111253489A-8FM.gif)
[0027] 以上为第一轮PCR反应体系,以第一轮PCR产物为模板做第二轮PCR。
[0028](2)第二轮PCR反应体系:
[0029] PCR各个成分的用量:引物浓度为1OD溶于400ul ddH2O。
[0030] 上游引物-1:2ul、下游引物-28:2ul、第一轮pcr产物:1ul、dNTP:1ul(25mM each)、10X pfu Buffer:5ul、Pfu:0.4ul(5u/ul)、ddH2O补齐至50ul。
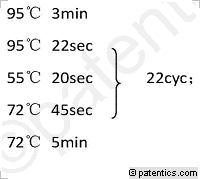
[0031](3)第二轮PCR程序:
[0032] /CN111253489A/CN111253489A-9FM.gif)
[0033] 以第二轮PCR琼脂糖凝胶电泳,回收纯化好的片段备用酶切。
[0034] 进一步,所述第一步的换载酶切将PCR产物进行酶切。PCR产物片段酶切体系50ul、纯化回收好的片段:1ug(20ul)、10X FD Buffer:5ul、EcoRI:1ul(10u/ul)、HindIII:1ul(10u/ul)、ddH2O:23ul。
[0035] 所述第一步的载体的酶切体系:PCDNA3.1+:1ug、10X FD Buffer:5ul、EcoRI:1ul(10u/ul)、HindIII:1ul(10u/ul)、ddH2O:42ul。放入37℃恒温水浴锅中反应2h。回收酶切的载体和片段。
[0036] 所述第一步的目的片段与载体连接,将回收纯化好的目的DNA片段和载体,进行连接。连接体系:20ul、酶切目的片段:8ul、酶切载体PCDNA3.1+:4ul、10X T4 DNAligase Buffer:2ul、T4 DNAligase:1ul(5u/ul)、ddH20补充至:20ul。
[0037] 转化,筛选克隆:连接混合液放在22℃PCR仪1h即可。
[0038] 进一步,所述第三步的VHH文库片段的制备,电转化和文库构建。选用M13噬菌体展示系统展示VHH抗体文库,由pMECS噬菌粒载体、E.coli TG1和M13KO7辅助噬菌体组成。在噬菌粒载体pMECS中,Pst I酶切位点之前的序列为pelB分泌信号肽和抗体第一个框架区部分氨基酸的编码序列,pelB信号肽能够将后续多肽引导分泌至周质腔。Not I酶切位点之后是HA和6×His标签的编码序列,用于融合蛋白的纯化或检测。
[0039] 本发明的另一目的在于提供一种所述纳米抗体在结合人的靶点阻断信号通路中的应用。
[0040] 本发明的另一目的在于提供一种所述纳米抗体在制备肿瘤检测或治疗的试剂中的应用。
[0041] 如上所述纳米抗体在制备用于提高动物免疫水平的免疫佐剂或者有病毒和/或细菌传播时的免疫促进剂中的应用。
[0042] 综上所述,本发明的优点及积极效果为:本发明的TIM-3抗原为哺乳动物细胞瞬时转染表达,免疫动物使用羊驼,筛选使用生物素化方法,筛选得到的纳米抗体片段为自己独特的基因序列。抗体可以用于结合人的靶点阻断信号通路的结合,可以用于作为肿瘤治疗,肿瘤检测等。目的片段扩增,换载酶切,目的片段与载体连接,转化,筛选克隆;蛋白鉴定表达;构建抗体库:样品总RNA提取及cDNA合成,VHH文库片段的制备,电转化和文库构建;选用M13噬菌体展示系统展示VHH 抗体文库,由pMECS噬菌粒载体、E.coli TG1和M13KO7辅助噬菌体组成。在噬菌粒载体pMECS中,Pst I酶切位点之前的序列为pelB分泌信号肽和抗体第一个框架区部分氨基酸的编码序列,pelB信号肽能够将后续多肽引导分泌至周质腔;Not I酶切位点之后是HA和6×His标签的编码序列,可用于融合蛋白的纯化或检测。使用抗原对纳米抗体库进行多次筛选,获得特异性的纳米抗体噬菌体,测序得到目的片段。
[0043] 纳米抗体与抗原的结合位点与单克隆抗体不同,使用纳米抗体替代单克隆抗体可以提高或者协同增强与抗原的结合能力。纳米抗体并不具备完整的抗体结构,缺少Fc端以及Y型结构,致使纳米抗体不容易被识别,可以轻易逃避免疫系统的捕捉。
[0044] 本发明使用HEK293细胞株对抗原进行表达,使用哺乳动物表达系统表达人的蛋白可以最大化的保证蛋白的原始结构,保证了蛋白拥有翻译后修饰以及糖基化等真核蛋白特有的修饰,可以使获取的蛋白具有较高活性。该方法最大地的保证了蛋白的原始结构与活性。
[0045] 本发明使用羊驼作为免疫动物可以更好的保定以及节约抗原的用量。本方法筛选得到的纳米抗体可以高效、特异性的结合到靶点上。
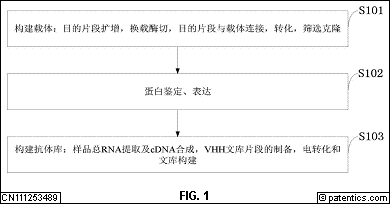
[0046] 图1
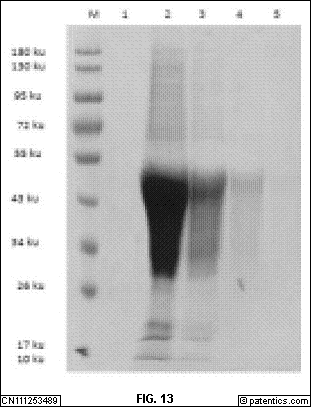
是本发明实施例提供的纳米抗体的筛选和鉴定方法流程图。
[0047] 图2
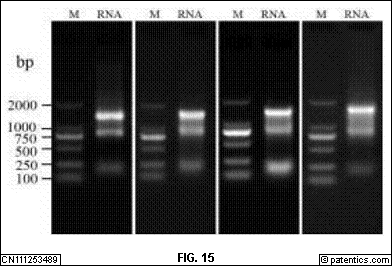
是本发明实施例提供的PCR琼脂糖凝胶电泳原图。
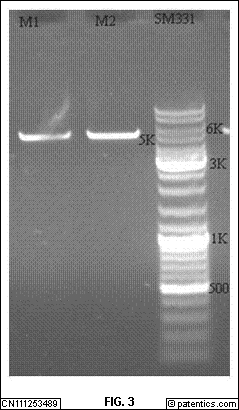
[0048] 图3
是本发明实施例提供的载体PCDNA3.1+切过后琼脂糖凝胶电泳图。
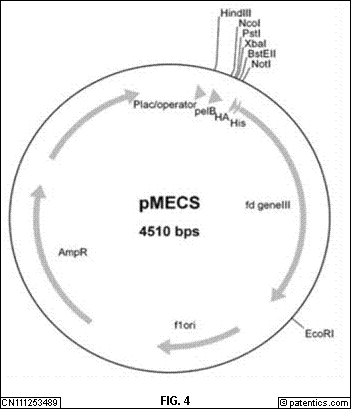
[0049] 图4
是本发明实施例提供的序列编码噬菌体PIII衣壳蛋白示意图。
[0050] 图5
是本发明实施例提供的噬菌粒pMECS质粒图谱。
[0051] 图6
是本发明实施例提供的总RNA样品凝胶电泳图。
[0052] 图7
是本发明实施例提供的PD-1PCR扩增结果示意图;
[0053] 图中:M.DL-10 000Marker;1.目的条带。
[0054] 图8
是本发明实施例提供的PD-L1 PCR扩增结果示意图;
[0055] 图中:M.DL-10 000Marker;1.目的条带。
[0056] 图9
是本发明实施例提供的TIM-3PCR扩增结果示意图;
[0057] 图中:M.DL-10 000Marker;1.目的条带。
[0058] 图10
是本发明实施例提供的CTLA-4PCR扩增结果示意图;
[0059] 图中:M.DL-10 000Marker;1.目的条带。
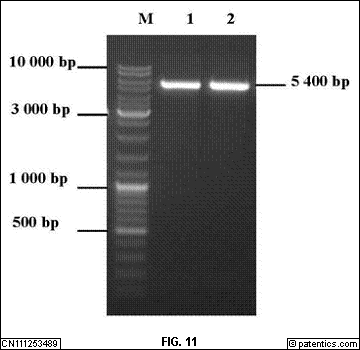
[0060] 图11
是本发明实施例提供的酶切鉴定结果示意图;
[0061] 图中:1,2.pcDNA3.1酶切条带;M.DL-10 000Marker。
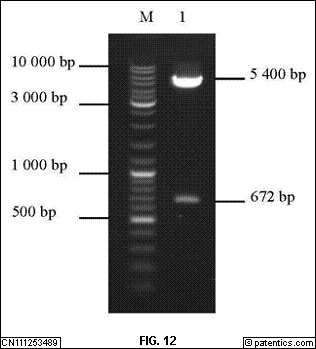
[0062] 图12
是本发明实施例提供的TIM-3酶切鉴定示意图。
[0063] 图13
是本发明实施例提供的TIM-3SDS-PAGE检测结果示意图。
[0064] 图14
是本发明实施例提供的TIM-3特异性WB检测示意图。
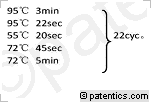
[0065] 图15
是本发明实施例提供的总RNA样品凝胶电泳图。
[0066] 图16
是本发明实施例提供的TIM-3骆驼抗体片段PCR扩增产物电泳分析示意图;
[0067] 图中:A,骆驼抗体片段1st PCR扩增;B,骆驼纳米抗体VHH片段2nd PCR扩增。
[0068] 图17
是本发明实施例提供的载体自连检测示意图。
[0069] 图18
是本发明实施例提供的连接小试抗性平板菌落计数示意图。
[0070] 图19
是本发明实施例提供的四种VHH文库的库容测定示意图。
[0071] 图20
是本发明实施例提供的菌落PCR产物琼脂糖凝胶电泳分析示意图。
[0072] 图21
是本发明实施例提供的筛选抗原SDS-PAGE和Western-blot检测示意图。
[0073] 图22
是本发明实施例提供的TIM-3生物素标记效率检测示意图。
[0074] 图23
是本发明实施例提供的TIM-3纳米抗体的细胞毒性示意图。图中,BHK-21:BHK-21细胞组;Kidney:绵羊肾细胞组;MDBK:MDBK细胞组。数据表示为平均值±SD。
[0075] 图24
是本发明实施例提供的TIM-3对免疫细胞的NO分泌水平影响示意图,图中,negative:阴性对照组;PBS:PBS 组;TIM-3:TIM-3纳米抗体组。
[0076] 为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0077] 针对现有的单克隆抗体存在异源性;单克隆抗体体积相对较大,单克隆抗体开发周期长,生产成本高,产量低;单克隆抗体类药物价格昂贵,研发复杂,人源化复杂,成功率有限;难以大规模生产;不稳定,容易降解,保存成本高;容易污染,维护成本高昂的问题。本发明的方法筛选得到的纳米抗体可以高效、特异性的结合到靶点上。
[0078] 下面结合附图对本发明的应用原理作详细的描述。
[0079] 本发明的TIM-3抗原为哺乳动物细胞瞬时转染表达,免疫动物使用羊驼,筛选使用生物素化方法,筛选得到的纳米抗体片段为自己独特的基因序列。
[0080] 本发明实施例的纳米抗体为TIM-3纳米抗体,其序列为SEQ ID NO:1。
[0081] 如图1
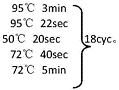
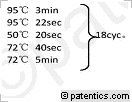
所示,本发明实施例提供的纳米抗体的筛选和鉴定方法包括以下步骤:
[0082] S101:构建载体:目的片段扩增,换载酶切,目的片段与载体连接,转化,筛选克隆。
[0083] S102:蛋白鉴定、表达。
[0084] S103:构建抗体库:样品总RNA提取及cDNA合成,VHH文库片段的制备,电转化和文库构建。
[0085] 在本发明的优选实施例中,步骤S101的目的片段扩增具体为:
[0086](1)设计合成引物2条,通过PCR的方法扩增出足够量的目的产物。引物的序列为:
[0087] SEQ ID NO:2TIM-3上游引物
[0088] 5′-GACACGAATTCGCCACCATGTTCAGCCACC-3′。
[0089] SEQ ID NO:3TIM-3下游引物
[0090] 5-GTGTCAAGCTTTCACTTGTCATCATCATCCTTGTA-3′。
[0091](2)PCR反应采用的是pfu高温聚合酶。
[0092] 在本发明的优选实施例中,步骤S101中的PCR各个成分的用量:引物浓度为1OD溶于400ul ddH2O。
[0093] 反应体系:50ul。引物mix,(1/28)0.4ul x 13.6共8ul。10X pfu Buffer:5ul。每段上下游引物各2ul。Pfu:0.4ul(5u/ul)。ddH2O分别补水至50ul。
[0094] 目的片段PCR的方法扩增具体步骤为:
[0095](1)第一轮PCR程序:
[0096] /CN111253489A/CN111253489A-10FM.gif)
[0097] 以上为第一轮PCR反应体系,以第一轮PCR产物为模板做第二轮PCR。
[0098](2)第二轮PCR反应体系:
[0099] PCR各个成分的用量:(引物浓度为1OD溶于400ul ddH2O)
[0100] 上游引物-1:2ul、下游引物-28:2ul、第一轮pcr产物:1ul、dNTP:1ul(25mM each)、10X pfu Buffer:5ul、Pfu:0.4ul(5u/ul)、ddH2O补齐至50ul。
[0101](3)第二轮PCR程序:
[0102] /CN111253489A/CN111253489A-11FM.gif)
[0103] 以第二轮PCR琼脂糖凝胶电泳,回收纯化好的片段备用酶切。
[0104] 换载酶切,将PCR产物进行酶切。
[0105] PCR产物片段酶切体系50ul、纯化回收好的片段:1ug(20ul)、10X FD Buffer:5ul、EcoRI:1ul(10u/ul)、HindIII:1ul(10u/ul)、ddH2O:23ul。
[0106] 以上体系放入37℃恒温水浴锅中反应2h。
[0107] 载体的酶切体系:
[0108] PCDNA3.1+:1ug、10X FD Buffer:5ul、EcoRI:1ul(10u/ul)、HindIII:1ul(10u/ul)、ddH2O:42ul。
[0109] 以上体系放入37℃恒温水浴锅中反应2h。回收酶切的载体和片段。
[0110] 目的片段与载体连接,将回收纯化好的目的DNA片段和载体,进行连接。
[0111] 连接体系:20ul、酶切目的片段:8ul、酶切载体PCDNA3.1+:4ul、10X T4 DNAligase Buffer:2ul、T4 DNAligase:1ul(5u/ul)、ddH20补充至:20ul。
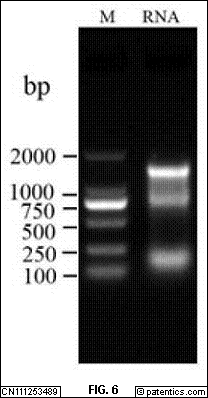
[0112] 转化,筛选克隆:连接混合液放在22℃PCR仪1h即可。
[0113] 下面结合具体实施例对本发明的应用原理作进一步的描述。
[0114] 实施例1
[0115] 本发明实施例提供的载体构建方法为:
[0116] 1、目的片段扩增具体为:
[0117](1)设计合成引物,通过PCR的方法扩增出足够量的目的产物。
[0118](2)PCR反应采用的是pfu高温聚合酶。
[0119] PCR各个成分的用量:(引物浓度为10D溶于400ul ddH2O)
[0120] 反应体系:50ul。引物mix,(1/28)0.4ul x 13.6共8ul。10X pfu Buffer,5ul。每段上下游引物,各2ul。Pfu,0.4ul(5u/ul)。ddH2O,分别补水至50ul。
[0121] 目的片段PCR的方法扩增具体步骤为:
[0122](1)第一轮PCR程序:
[0123] /CN111253489A/CN111253489A-12FM.gif)
[0124] 以上为第一轮PCR反应体系,以第一轮}PCR产物为模板做第二轮PCR。
[0125](2)第二轮PCR反应体系:
[0126] PCR各个成分的用量:(引物浓度为10D溶于400ul ddH2O)。
[0127] /CN111253489A/CN111253489A-13FM.gif)
[0128](3)第二轮PCR程序:
[0129] /CN111253489A/CN111253489A-14FM.gif)
回收纯化好的片段备用酶切。
[0130] 2、换载酶切,将上述PCR产物进行酶切。
[0131] /CN111253489A/CN111253489A-15FM.gif)
[0132] 以上体系放入37℃恒温水浴锅中反应2h。
[0133] 载体的酶切体系:
[0134] /CN111253489A/CN111253489A-16FM.gif)
[0135] 在本发明实施例中,如图3
所示,本发明实施例提供的载体PCDNA3.1+切过后琼脂糖凝胶电泳图。以上体系放入37℃恒温水浴锅中反应2h。回收酶切的载体和片段。
[0136] 3、目的片段与载体连接,将回收纯化好的目的DNA片段和载体,进行连接。
[0137] /CN111253489A/CN111253489A-17FM.gif)
[0138] 4、转化,筛选克隆:上述连接混合液放在22℃PCR仪1h即可。
[0139] 实施例2。
[0140] 本发明实施例提供的纳米抗体库构建具体为:
[0141] 1)实验设计
[0142] 选用M13噬菌体展示系统展示VHH抗体文库,该系统由pMECS噬菌粒载体、E.coli TG1和M13KO7辅助噬菌体组成。在噬菌粒载体pMECS中,Pst I酶切位点之前的序列为pelB分泌信号肽和抗体第一个框架区部分氨基酸的编码序列,pelB信号肽能够将后续多肽引导分泌至周质腔。Not I酶切位点之后是HA和6×His标签的编码序列,可用于融合蛋白的纯化或检测。紧随其后的序列编码噬菌体PIII衣壳蛋白(图4
所示)。6×His标签和gene III序列之间有一个琥珀终止密码子,在琥珀终止密码子抑制型菌株(例如E.coli TG1)中有10%~20%的琥珀终止密码子能够翻译为谷氨酸(Glu,或E),VHH与gene III蛋白融合表达,当利用辅助噬菌体M13KO7拯救后,VHH抗体展示在噬菌体尾部PIII蛋白的N末端。
[0143] 在本发明实施例中,如图4
所示,本发明实施例提供的序列编码噬菌体PIII衣壳蛋白示意图。
[0144] 因此,首先抽提样品的总RNA,并反转录为cDNA,然后利用CAL-leader和CAL-CH2引物对,扩增骆驼抗体片段。切胶回收上述PCR的~600bp的条带,作为后续PCR的模板,使用VHH-back和PMCF引物扩增VHH基因片段。在VHH基因片段的3’端引入Not I酶切位点(VHH片段的5’端带有Pst I酶切位点),并利用酶切和连接反应,将该片段插入pMECS噬菌粒载体,然后转化到大肠杆菌TG1,构建M13单链丝状噬菌体展示骆驼纳米抗体免疫文库。
[0145] 2)实验材料
[0146](1)骆驼外周血淋巴细胞样品采集自骆驼外周血。
[0147](2)cDNA合成、PCR扩增、限制性内切酶、T4 DNA连接酶等试剂盒和工具酶主要购自Thermo Scientific和New England Biolabs等公司。
[0148](3)pMECS、E.coli TG1、Helper phage M13KO7等实验材料由人畜共患病实验室保存。
[0149] 如图5
所示,本发明实施例提供的噬菌粒pMECS质粒图谱。
[0150] 3)实验结果
[0151](1)样品总RNA提取及cDNA合成
[0152] 使用Trizol试剂分别抽提骆驼外周血淋巴细胞样品总RNA,通过琼脂糖凝胶电泳检测总RNA的质量。
[0153] 如图6
所示,本发明实施例提供的总RNA样品凝胶电泳图。
[0154] 图中,M为DL2000 DNA marker。
[0155] 总RNA样品存在非常轻微的降解现象,28S、18S及5S rRNA条带均清晰可见,且28S条带亮度大于18S,表明RNA的完整性较好。Nanodrop测定RNA样品的浓度,结果表明RNA样品浓度及纯度符合要求(表1)。以10μg总RNA为模板,反转录合成cDNA。
[0156] 表1:总RNA样品浓度及纯度
[0157] /CN111253489A/CN111253489A-18FM.gif)
[0158](2)VHH文库片段的制备
[0159] 分别以上述的cDNA为模板,使用引物CAL-leader和CAL-CH2扩增骆驼抗体片段,取适量PCR产物进行1%琼脂糖凝胶电泳检测,结果如图3A
所示:样品均有两条带,主带分子量大小约为600bp,另外在900bp处有一条非目标条带(此条带应为传统抗体的扩增片段)。将足量的PCR产物进行电泳,切胶回收600bp的主条带,作为后续PCR的模板,使用VHH-back 和PMCF引物扩增VHH。PCR扩增得到了分子量大小与预期一致(约为400bp)的目的条带。
[0160] 实施例3。
[0161] 本发明实施例提供的蛋白鉴定、表达,具体为:
[0162](一)构建哺乳动物细胞表达载体(质粒模板,C端加入His-Flag tag)
[0163] 1、扩增并抽提含有目的基因的载体质粒。
[0164] 2、亚克隆到真核表达载体pcDNA3.1中。
[0165] 3、测序验证构建质粒的准确性。
[0166] 4、通过中抽获得重组质粒pcDNA3.1。
[0167](二)哺乳动物细胞培养,蛋白表达和纯化小试
[0168] 1、细胞株及材料
[0169] 细胞株:HEK293细胞。
[0170] 培养基:DMEM(10%血清),DMEM(无血清)。
[0171] 培养器皿:10cm dish or 15cm dish
[0172] 2、HEK293细胞转染(10cm dish)
[0173](1)转染前24h以4-5X106/10cm培养皿的细胞总量铺板,当细胞生长状态良好且贴壁细胞密度达到50-80%时即可进行转染。
[0174](2)在1.5ml离心管中加入待转质粒DNA 5ug,混匀。
[0175](3)将10ul脂质体加入500ul DMEM培养液中,加入上述DNA,轻轻混匀,RT温育30min。
[0176](4)将含有DNA和脂质体的液体小心加入到培养皿中,分散均匀,置于37℃5%CO2的培养箱中培养72h。
[0177] 3、观察并收集细胞
[0178](1)转染72h后,小心吸干细胞培养液,用预冷的PBS润洗贴壁细胞1次。
[0179](2)离心收集细胞沉淀。
[0180] 4、裂解细胞
[0181](1)加细胞裂解液Lysis Buffer:50mM Tris(pH8.0),300mM NaCl,1%Triton X-100,1mM DTT,5%甘油。
[0182](2)200W冰浴超声10min。
[0183](3)16000rpmX20min,4℃,收集裂解上清液。
[0184] 5、Flag标签纯化
[0185](1)使用Flag填料,1ml柱子用于纯化。
[0186](2)柱子事先用Binding Buffer:(50mM Tris(pH 8.0),300mM NaCl,0.1%Triton X-100,1mM DTT,5%甘油)平衡10CV。
[0187](3)裂解后的细胞上清液加入到平衡好的柱子上。
[0188](4)样品上完,用Binding Buffer清洗柱子。
[0189](5)用Wash Buffer:(50mM Tris(pH8.0),500mM NaCl,1mM DTT,5%甘油)清洗柱子5-10CV,洗去若结合的非特异吸附的杂质。
[0190](6)用Elution Buffer:(50mM Tris(pH8.0),150mM NaCl,150μg/μl Flag peptide,1mM DTT,10%甘油)洗脱目标蛋白,收集目标蛋白。
[0191] 6、Western-blot检测
[0192](1)溶液准备:
[0193] 转印缓冲液:0.025M Tris base,0.192M甘氨酸,30%甲醇10×TBST:250mM Tris-HCl(pH 8.0),1.25M NaCl,0.5%Tween20。封闭液:1×TBST,3%脱脂奶粉。洗涤液:1×TBST。
[0194](2)实验程序:
[0195] A.膜准备:将PVDF膜切成条浸在甲醇中,室温条件下在摇床上摇1min,去除甲醇后加入1×TBST。
[0196] B.膜转印:
[0197] i.SDS-PAGE电泳。
[0198] ii.用夹心法电转至PVDF膜。
[0199] iii.海绵和滤纸浸泡在转印缓冲液预湿润。
[0200] iv.300mA转印80min,封闭液室温封闭1h或者4℃过夜。
[0201] C.抗体检测:
[0202] i.将一抗anti-Flag tag按说明书稀释,4℃孵育过夜。
[0203] ii.用洗涤液洗涤3次,每次5min。
[0204] iii.用封闭液来稀释二抗,室温下培育1h。
[0205] iv.用洗涤液洗涤3次,每次5min。
[0206] v.TMB显色检测。
[0207](三)蛋白鉴定结果:
[0208](1)由于每个蛋白具体的氨基酸组成和排列不相同,即使是分子量很接近的蛋白,SDS-PAGE胶上的表现也会有所不同。蛋白分子量marker是一个分子量的参考,在SDS-PAGE电泳上,目的蛋白有可能和在理论分子量完全吻合,也有可能高出或者低些。
[0209](2)分泌蛋白样品,有糖基化修饰,分子量常规会高于理论分子量。另外,蛋白的糖基化修饰过程不是完全均一的,经常会看见目的蛋白出现蛋白条带弥散,出现相互靠近的多条条带,这些是分泌蛋白的典型电泳表现。
[0210] 下面结合实验对本发明作进一步的描述。
[0211] 实验一
[0212] 一、实验蛋白表达与羊驼免疫
[0213] 试验采用PCR方法扩增TIM-3胞外区序列,构建重组pcDNA3.1质粒,将质粒转染至HEK293细胞株中表达目的蛋白,并对目的蛋白进行SDS-PAGE及Western-blot检测。结果表明:PCR法成功扩增出目的条带,1%琼脂糖凝胶电泳显示酶切成功。10%SDS-PAGE检测显示目的蛋白成功表达。Western-blot检测证明蛋白具有特异性。
[0214] 1材料与方法
[0215] 1.1材料
[0216] 1.1.1质粒与菌株
[0217] pcDNA3.1质粒、HEK293细胞和HEK293感受态细胞,均由新疆民族与地方高发病省部共建实验室保存。
[0218] 1.1.2主要生化试剂
[0219] 胎牛血清、DMEM培养基,购自Gibco公司。EcoRⅠ内切酶、HindⅢ内切酶、鼠抗His单克隆IgG抗体、辣根过氧化物酶(HRP)标记的兔抗鼠IgG,均购自生工生物工程(上海)股份有限公司。镍柱,购自GE公司。TMB显色液,购自北京中杉金桥生物技术有限公司。SM331 GeneRuler DNA Ladder Mix(Thermo Scientific公司)。(其余试剂均为sangon公司生产)。所用的酶为Thermo Scientific公司生产的NdeI,XhoI酶及对应的FDBuffer。电泳仪为北京六一仪器厂的DYY85型。PCR 产物纯化使用生工产的PCR纯化试剂盒.10X T4 DNAligase Buffer所用的连接酶为Thermo Thermo Scientific公司生产。
[0220] 1.2方法
[0221] TIM-3免疫原的制备
[0222] 通过Uniprot数据库获取淋序列,使应用Primer Premier 5.0软件设计引物,由生工生物工程(上海)股份有限公司合成。由生工生物工程(上海)股份有限公司合成,以合成片段为PCR模板。
[0223] 表1
[0224] /CN111253489A/CN111253489A-19FM.gif)
[0225] PCR各个成分的用量:(引物浓度为1OD溶于400ul ddH2O)
[0226] 表2
[0227] /CN111253489A/CN111253489A-20FM.gif)
[0228] TIM-3目的片段PCR程序:
[0229] 表3
[0230] /CN111253489A/CN111253489A-21FM.gif)
[0231] PCR完成后进行1%琼脂糖凝胶电泳,回收纯化好的片段酶切备用。
[0232] 酶切与鉴定
[0233] 将上述PCR产物进行酶切,PCR产物片段酶切体系50ul。
[0234] 表4
[0235] /CN111253489A/CN111253489A-22FM.gif)
[0236] /CN111253489A/CN111253489A-23FM.gif)
[0237] 以上体系放入37℃恒温水浴锅中反应2h。载体的酶切体系:
[0238] 表5
[0239] /CN111253489A/CN111253489A-24FM.gif)
[0240] 以上体系放入37℃恒温水浴锅中反应2h,回收酶切的载体和片段。目的片段与载体连接将回收纯化好的目的DNA片段和载体,进行连接。
[0241] 表6
[0242] /CN111253489A/CN111253489A-25FM.gif)
[0243] 将连接混合液置22℃PCR仪中作用1h。将上述连接液采用42℃热激法转入HEK293感受态细胞中,检测筛选出的阳性克隆,提取重组质粒。
[0244] SDS-PAGE检测
[0245] 转染HEK293细胞前24h铺板,当细胞生长状态良好且贴壁细胞密度达到50%~80%时进行转染。将10μL脂质体加到500μL DMEM培养基中,加入重组质粒5μg,轻轻混匀,常温温育30min。将含DNA和脂质体的液体小心加到培养皿中,分散均匀,置37℃、5%CO2培养箱中培养72h。吸干细胞培养液,离心收集细胞,加入细胞裂解液,200W冰浴超声10min。16 000r/min离心20min。收集裂解上清液进行10%SDS-PAGE电泳,确定蛋白表达之后使用镍柱纯化蛋白。
[0246] 特异性Western-blot检测
[0247] 将PVDF膜切成条浸在甲醇中,室温条件下在摇床中培养1min。去除甲醇后加入1×TBST进行SDS-PAGE电泳,用夹心法将蛋白电转移至PVDF膜。将滤纸浸泡在转印缓冲液预湿润后,300mA转印80min。于室温条件下用封闭液封闭1h。将一抗进行3 000倍稀释,4℃孵育过夜。用PBST洗涤3次,每次5min。然后用封闭液5000倍稀释二抗,室温条件下温育1h。用PBST洗涤3次,每次5min,使用TMB显色液显色。TIM3一抗为Anti-TIM3抗体(F38-2E2)(小鼠单克隆抗体F38-2E2 to TIM3,Anti-TIM3 antibody(F38-2E2)ab104709),二抗为山羊多克隆抗体二抗to小鼠IgG-H&L(HRP)抗体(abcam公司ab6789)。方法同上。
[0248] 初次免疫,使用弗氏完全佐剂0.5ml+蛋白0.5ml乳化,进行皮下与皮内注射免疫。弗氏不完全佐剂0.25ml+蛋白0.25ml 乳化均匀,分别在28天(二免)、49天(三免)、70天(四免)皮下免疫羊驼。弗氏不完全佐剂0.125ml+蛋白0.125ml乳化均匀,分别在91天(五免)、112天(六免)免疫羊驼。在第122天,分离淋巴细胞。
[0249] 表7
[0250] /CN111253489A/CN111253489A-26FM.gif)
[0251] 表8
[0252] /CN111253489A/CN111253489A-27FM.gif)
[0253] ELISA检测实验步骤
[0254] 抗原分别200ng/well,4度包被过夜。PBST(0.1%)洗板1次,1×bloker 300μl/well,37度封闭2h。PBST(0.1%)洗板1 次,0.5×blocker梯度稀释抗血清,100μl/wel加入板中,37度1h。PBST(0.1%)洗板3次,抗羊驼二抗用0.5×blocker稀释1:15000,100μl/孔加入板中37度1h。PBST(0.1%)洗板3次,PBS洗板3次,100μl/孔TMB显色约20min,50μl/孔2M H2SO4终止。酶标仪OD450-OD630nm读数。
[0255] 淋巴细胞分离:
[0256] 将淋巴细胞分离液预热至22℃。使用肝素钠抗凝管每只羊驼采集外周血200mL。使用等体积的组织稀释液稀释全血。在离心管中加入等体积的分离液。室温,水平转子1000g,离心30min。吸取白膜层,加入10mL PBS洗涤液洗涤白膜层细胞。250g,离心10min。弃上清,5mL的PBS重悬细胞,250g,离心10min。弃上清,5mL的PBS重悬细胞,250g,离心10min。弃上清,细胞使用Trizol重悬。
[0257] 二、骆驼纳米抗体免疫库的构建
[0258] 抽提骆驼外周血淋巴细胞总RNA,并反转为cDNA。利用骆驼抗体引物对,扩增出VHH文库片段。将四种VHH文库片段分别插入pMECS噬菌粒载体,并转化到大肠杆菌TG1,构建噬菌体展示抗体免疫文库,库容>109。从文库中随机挑取20 个克隆,进行序列测定和分析,确保文库中99%以上的克隆包含了目的插入序列。
[0259] 1.实验材料
[0260] cDNA合成、PCR扩增、限制性内切酶、T4 DNA连接酶等试剂盒和工具酶主要购自Thermo Scientific和New England Biolabs 等公司。pMECS、E.coli TG1、Helper phage M13KO7等实验材料由实验室保存。
[0261] 2.实验方法
[0262] 样品总RNA提取及cDNA合成
[0263] 使用Trizol抽提骆驼外周血淋巴细胞样品的总RNA,通过琼脂糖凝胶电泳检测总RNA的质量。
[0264] VHH文库片段的制备
[0265] 分别以cDNA为模板,使用引物CAL-leader和CAL-CH2扩增骆驼抗体片段,取适量PCR产物进行1%琼脂糖凝胶电泳检测。
[0266] 引物CAL-leader序列:GTCCTGGCTGCTCTTCTACAAGG
[0267] 引物CA-CH2序列:GGTACGTGCTGTTGAACTGTTCC
[0268] PCR体系:
[0269] 使用pfu高保真DNA聚合酶(TransStart FastPfu DNA Polymerase,AP221-01)。
[0270] /CN111253489A/CN111253489A-28FM.gif)
[0271] PCR程序:
[0272] /CN111253489A/CN111253489A-29FM.gif)
[0273] 连接和转化预实验
[0274] 在正式建库之前,通过连接和转化预实验,对噬菌粒载体质量、载体和VHH文库片段的连接效率进行测试和评估。使用T4 DNA ligase将经Pst I/Not I酶切后的VHH片段和同样经Pst I/NotI酶切后的噬菌粒载体pMECS以不同的比例进行连接反应(每组的载体用量相同),然后转化E.coli TG1化学感受态细胞,涂布氨苄抗性平板进行菌落计数。
[0275] 样品有两条带,主带分子量大小约为600bp,另外在900bp处有一条非目标条带,此条带应为传统抗体的扩增片段。将足量的PCR产物进行电泳,切胶回收600bp的主条带,作为后续PCR的模板,使用VHH-back和PMCF引物扩增VHH,PCR 扩增得到了分子量大小与预期一致,约为400bp的目的条带。
[0276] 引物VHH-back序列:GATGTGCAGCTGCAGGAGTCTGGRGGAGG
[0277] 引物PMCF序列:CTAGTGCGGCCGCTGAGGAGACGGTGACCTGGGT。
[0278] PCR体系:
[0279] /CN111253489A/CN111253489A-30FM.gif)
[0280] PCR程序:
[0281] /CN111253489A/CN111253489A-31FM.gif)
[0282] 电转化和文库构建
[0283] 在正式建库的连接实验中,按照连接和转化预实验得出的最佳比例,将载体与四种VHH片段进行连接反应。纯化后的连接产物经电转化到大肠杆菌TG1,得到了15ml转化产物。取10μl(即为10-2ml)进行一系列10倍梯度稀释,并取10-4、10-5和10-6共三个梯度进行涂氨苄抗性平板计数,以评估文库库容,库容=克隆数×稀释倍数×转化产物总体积(ml)。将剩下的转化产物全部涂布到15块直径为15cm的Amp抗性平板培养过夜,次日分别刮下来混匀后,加入20%终浓度的甘油,分装并冻存于-80℃。
[0284] 电转化实验方案:
[0285] 1)制备E.coli TG1电转化感受态细胞。
[0286] 2)将纯化后的连接产物加入适量TG1感受态细胞,混合均匀后,分装至0.2cm电转化杯。
[0287] 3)利用电转化仪进行电转化,BIORAD推荐转化条件:2.5kV,25μF,200Ω。
[0288] 4)加入2YT培养基至电转化杯中,重悬感受态细胞,37℃,150rpm复苏30min。
[0289] 免疫文库质量分析
[0290] 从每种文库的梯度稀释平皿上随机挑取20个单克隆,使用引物MP57和PMCF进行菌落PCR。使用引物MP57 (-TTATGCTTCCGGCTCGTATG-)对这些克隆进行测序。
[0291] PCR体系:
[0292] 使用Taq DNA聚合酶(2×Taq Plus MasterMix,CW2849M)。
[0293] /CN111253489A/CN111253489A-32FM.gif)
[0294] PCR程序:
[0295] /CN111253489A/CN111253489A-33FM.gif)
[0296] 2结果
[0297] 2.1 PCR扩增
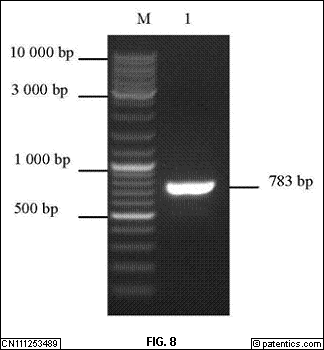
[0298] 经PCR扩增得到大小相符的目的条带,用1%琼脂糖凝胶电泳检测目的条带清晰,与预期大小相符,说明成功扩增出目的条带。图7
是本发明实施例提供的PD-1PCR扩增结果示意图;图中:M.DL-10 000Marker;1.目的条带。图8
是本发明实施例提供的PD-L1 PCR扩增结果示意图;图中:M.DL-10 000Marker;1.目的条带。图9
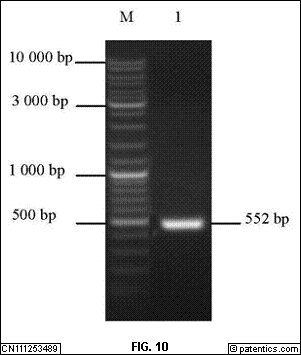
是本发明实施例提供的TIM-3PCR扩增结果示意图;图中:M.DL-10 000Marker;1.目的条带。图10
是本发明实施例提供的CTLA-4PCR扩增结果示意图;图中:M.DL-10 000Marker;1.目的条带。
[0299] 图11
是本发明实施例提供的酶切鉴定结果示意图;图中:1,2.pcDNA3.1酶切条带;M.DL-10 000Marker。
[0300] 2.2酶切鉴
[0301] pcDNA3.1质粒酶切后用1%琼脂糖凝胶电泳检测,得到大小为为5 300bp的条带(图10
)与预期大小相符,说明回收片段酶切成功。
[0302] 回收酶切的载体和片段进行凝胶电泳验证。使用质粒小提试剂盒提取目的片段。
[0303] TIM-3酶切鉴定,如图12
所示。
[0304] 2.3 SDS-PAGE检测
[0305] TIM-3 SDS-PAGE检测结果,如图13
所示。
[0306] 细胞培养液经10%SDS-PAGE凝胶电泳分析显示,在ku处出现目的条带,与预期大小相符,证明蛋白成功表达,同时证明该蛋白以可溶性形式表达。特异性Western-blot检测,如图14
所示。
[0307] 羊驼免疫实验结果,见表9
[0308] 表9
[0309] 12800倍 25600倍 51200倍 102400倍 204800倍 409600倍 PBS
TIM-3免疫前 0.107 0.075 0.055 0.045 / / 0.042
TIM-3六免 OUT OUT OUT 1.813 0.983 0.47 0.04
[0310] 噬菌体文库的构建
[0311] 样品总RNA提取及cDNA合成
[0312] 结果如图15
所示,四种总RNA样品存在非常轻微的降解现象,28S、18S及5S rRNA条带均清晰可见,且28S条带亮度大于18S,表明RNA的完整性较好。Nanodrop测定RNA样品的浓度,结果表明RNA样品浓度及纯度符合要求(表10)。每个项目以10μg总RNA为模板,反转录合成cDNA。
[0313] 表10总RNA样品浓度及纯度
[0314] /CN111253489A/CN111253489A-34FM.gif)
[0315] VHH文库片段的制备
[0316] 结果如图16
-A所示:四种样品均有两条带,主带分子量大小约为600bp,另外在900bp处有一条非目标条带(此条带应为传统抗体的扩增片段)。将足量的PCR产物进行电泳,切胶回收600bp的主条带,作为后续PCR的模板,使用VHH-back 和PMCF引物扩增VHH。结果如图16
-B所示:PCR扩增得到了分子量大小与预期一致(约为400bp)的目的条带。
[0317] 结果如图17
和图18
所示:Pretest 1无插入片段,即载体自连,有9个菌落。载体与VHH基因片段以不同的比例连接和转化到E.coli TG1,在氨苄抗性平板上的菌落生长情况。转化后加入培养基至1ml进行复苏,取100μL涂布到平皿上,进行培养和计数,故平板上的克隆数目为实际的十分之一。
[0318] 电转化和文库构建
[0319] 如图19
所示,TIM-3文库-5和-6梯度的克隆数量分别为~2000和256个,故库容为:[256×15×106+(2000+256) ×15×105]÷2=3.6×109。
[0320] 免疫文库质量分析
[0321] 结果如图20
所示,所有的克隆都有分子量大小约为500bp的特异性条带,表明这些克隆均为阳性。使用引物MP57 (TTATGCTTCCGGCTCGTATG)对这些克隆进行测序。
[0322] 三、实验纳米抗体的筛选
[0323] 1、实验材料:NeutrAvidin预包被的板子、Dynabeads和试剂主要购自Thermo Scientific和国药等公司。Helper phage、E.coli TG1等实验材料由实验室保存。
[0324] 2、实验方法
[0325] TIM-3抗血清效价检测,用梯度稀释ELISA方法检测TIM-3免疫前后血清效价,包被TIM-3(200ng/well),加入梯度稀释抗血清,二抗为抗羊驼-HRP 1:15000稀释使用,最后用TMB底物显色。
[0326] TIM-3蛋白样品SDS-PAGE、Western-blot和生物素标记,SDS-PAGE检测TIM-3蛋白上样量为2μg,Western-blot检测,抗血清1:20000稀释,抗羊驼-HRP二抗工作浓度为1:2000,用化学发光法显色。方法同上。
[0327] 靶点TIM-3的生物素标记及效率检测,对TIM-3进行生物素标记,条件为0.25mg/ml,pH 7.4,蛋白与生物素比例为1:15,室温标记1h。标记好的蛋白,采用PD-Midi脱盐柱去除游离生物素,置换buffer为PBS 5%甘油pH7.4,最后各分装后保存于-70℃。为了检测TIM-3的生物素标记效率,取两份1.5μg标记后的b-TIM-3蛋白,然后分别加入5μg链霉亲和素(SA)和5μl PBS。另外同样取5μg SA,加入5μl PBS作为SA样品对照。室温反应1h后,各加入5μl的非还原loading buffer,不加热变性,直接进行SDS-PAGE。
[0328] 体外定向筛选,用构建好的免疫文库,针对TIM-3进行3轮筛选。
[0329] 鉴定
[0330] 1)Monoclonal phage ELISA analysis
[0331] 将第二、三轮洗脱的富集产物中挑322个克隆进行Monoclonal phage ELISA验证,分别包被TIM-3和BSA对照200ng/well。
[0332] 2)Soluble ELISA analysis
[0333] 将TIM-3序列独特克隆(in E.coli TG1)在30℃进行IPTG诱导表达,离心后收集菌体,进行周质腔抽提。将周质腔抽提样品用0.5×blocker稀释10倍加入包被并封闭好的TIM-3和BSA中,同时设置TG1(不含phagemid)周质腔抽提物作为阴性对照。使用mouse anti-HA tag单克隆抗体(ProteinTech,1:5000稀释)作二抗,羊抗鼠-HRP(1:5000稀释)作为三抗,检测可溶性表达纳米抗体的活性。
[0334] 构建pET28a-SUMO载体表达和纯化为提高纳米抗体的表达量和活性,将11个有活的纳米抗体TIM-3克隆构建到pET28a-SUMO载体中,进行胞内表达,超声破菌后用Ni柱纯化。
[0335] 将纯化的纳米抗体用0.5×blocker梯度稀释加入包被并封闭好的TIM-3和BSA(200ng/well)中,同时设置PBS作为阴性对照。使用mouse anti-HA tag单克隆抗体(ProteinTech,1:5000稀释)作为二抗,羊抗鼠-HRP(1:5000稀释)作为三抗,检测可溶性纯化纳米抗体的活性。
[0336] 3、实验结果
[0337] TIM-3项目抗血清效价检测结果抗血清效价高,达到1:204800。
[0338] 表11羊驼免疫前后血清效价检测
[0339] 抗血清稀释倍数 免疫后血清 免疫前血清
200 / 0.76
400 OUT 0.725
800 OUT 0.547
1600 OUT 0.35
3200 OUT 0.232
6400 OUT 0.163
12800 OUT 0.107
25600 OUT 0.075
51200 OUT 0.055
102400 1.813 0.045
204800 0.983 /
409600 0.47 /
PBS 0.04 0.042
[0340] OUT代表OD450nm下的ELISA值大于3。
[0341] TIM-3蛋白样品SDS-PAGE、Western-blot和生物素标记
[0342] 1)筛选抗原SDS-PAGE和Western-blot检测,如图21
所示。TIM-3经SDS-PAGE检测,结果如图21
显示,蛋白纯度高,无降解,分子量约45kDa,满足后续标记和筛选要求。Western-blot检测结果抗血清特异识别TIM-3。
[0343] 2)TIM-3生物素标记效率检测
[0344] 生物素标记效率检测结果如图22
所示,SA+b-TIM-3泳道比SA+PBS有明显条带迁移,同时在45kDa附近的b-TIM-3条带较等量的b-TIM-3+PBS组有明显减少,几乎没有蛋白残留,因此估计标记效率为>90%。
[0345] 体外定向筛选
[0346] 用构建好的免疫文库,针对b-TIM-3进行3轮筛选,策略和结果如下表:
[0347] 表12
[0348] /CN111253489A/CN111253489A-35FM.gif)
[0349] /CN111253489A/CN111253489A-36FM.gif)
[0350] 筛选结果显示,在第二轮筛选时出现明显富集,在第三轮有一定程度的富集。
[0351] 鉴定
[0352] 1)Monoclonal phage ELISA analysis
[0353] ELISA检测结果附件(TIM-3monoclonal phage ELISA汇总),阳性率约80%,挑选其中51个强阳性克隆测序,结果共有23个独特序列克隆。
[0354] 2)Soluble ELISA analysis
[0355] 结果见表11,共有11个纳米抗体能可溶性表达且有较好活性。相同的背景色标记的克隆的序列相似性较高。
[0356] 表13
[0357] /CN111253489A/CN111253489A-37FM.gif)
[0358] 下面结合具体检测试验对筛选的纳米抗体进行检测:
[0359] 1方法
[0360] 1.1纳米抗体细胞毒性实验
[0361] 37℃温水复苏BHK-21细胞、MDBK细胞、羊的肾细胞,加入细胞培养液(90%DMEM+10%FBS),传代到96孔板,6×103个细胞每孔。等待细胞贴壁,并孵育3小时。加入TIM-3纳米抗体,终浓度梯度设置为:5μg/ml,10μg/ml,20μg/ml,40 μg/ml。共4个梯度。将20μl/孔MTS试剂加入每个孔中37℃温育3小时。振荡后在492nm测量吸光度。
[0362] 1.2纳米抗体与噬菌体的NO检测实验
[0363] 将小鼠免疫细胞加入96孔板3h后加入终浓度为1μg/mL的T7噬菌体,然后孵育24h。将TIM-3纳米抗体使用培养液(90%DMEM+10%FBS)稀释加入孔中,终浓度调整为10μg/mL,20μg/mL,40μg/mL,80μg/mL,每个样品三个复孔。细胞培养箱中培养细胞38h h。对照组加入提取液100μL,试剂一50μL,试剂二50μL(均为购自北京索莱宝生物技术有限公司NO水平检测试剂盒中的试剂).测定实验组加入样品100μL,试剂一50μL,试剂二50μL。混匀然后室温静置15min,测定550nm处吸光值。
[0364] 1.3数据分析
[0365] 结果表示为平均值±标准偏差(SD)。使用GraphPad Prism 8软件进行统计学分析通过Mann-Whitney U检验进行差异显著分析(*=P<0.05,**=P<0.01)。
[0366] 1.4纳米抗体注射小鼠后,小鼠体内IL-4、IFN-γ细胞因子以及NO分泌变化水平
[0367] 注射抗体BalB/C小鼠,每只0.1mg。每只注射0.2ml,浓度为0.5mg/mL。分组如下:(例如PBS对照组有两组,并非重复与错误,后续会用金黄色葡萄球菌与无乳链球菌分别攻毒,故每个抗体注射分了两组)。
[0368] /CN111253489A/CN111253489A-38FM.gif)
[0369] 免疫三天后,采血并分离血清,检测细胞因子IL-4、IFN-γ与NO水平(IL-4检测试剂盒、IFN-γ检测试剂盒与NO水平检测试剂盒均购自北京索莱宝生物技术有限公司)。
[0370] 1.5金黄色葡萄球菌和无乳链球菌攻毒保护实验
[0371] 然后分别注射金黄色葡萄球菌与无乳练球菌,其中金黄色葡萄球菌注射150μl/1.9×109cfu(多次研究判定的小鼠最低致死剂量)。无乳练球菌注射无乳200μl/5.1×1010cfu(多次研究判定的小鼠最低致死剂量)。24小时后观察小鼠状态并记录。
[0372] 2结果
[0373] 2.1纳米抗体的细胞毒性测定,
[0374] 如图23T
IM-3纳米抗体的细胞毒性所示。图24
中,BHK-21:BHK-21细胞组;Kidney:绵羊肾细胞组;MDBK:MDBK细胞组。数据表示为平均值±SD。
[0375] 不同浓度的纳米抗体对小鼠(BHK-21细胞)、绵羊(绵羊肾细胞)以及牛的细胞(MDBK细胞)均没有细胞毒性。证明TIM-3纳米抗体对动物细胞没有细胞毒性。
[0376] MTT商品名称噻唑蓝通过特定波长的分光光度测定,可对细胞存活及生长情况进行定量测定和分析。BioVision的MTS细胞增殖分析试剂盒是一种比色法,是MTT的升级版,用于在增殖和细胞毒性分析中灵敏定量活细胞,可以用于判断试剂对细胞有无毒性。通过验证纳米抗体对哺乳动物细胞BHK-21,MDBK或绵羊肾细胞没有毒性,可以安全的用于动物。
[0377] 2.2NO激活实验测定,
[0378] 如图24T
IM-3对免疫细胞的NO分泌水平影响所示,图中,negative:阴性对照组;PBS:PBS组;TIM-3:TIM-3纳米抗体组。
[0379] 在免疫细胞添加T7噬菌体后,添加不同浓度的TIM-3纳米抗体共同孵育24h后,显示TIM-3纳米抗体对于NO浓度的增强与浓度呈正相关,TIM-3纳米抗体浓度越高,NO检测值越大,代表NO的分泌水平越高。TIM-3纳米抗体在10μg/mL、20μg/mL、40μg/mL时,增强作用较小,在第80μg/mL时达到最大值。
[0380] 一氧化氮(Nitric Oxide,NO)作为细胞间及细胞内的信息传递物质,发挥了信号传递的作用,是一种新型的生物信使分子。中国农业科学院兰州兽医研究所农业部兽医病原生物学国家重点实验室和动物病毒学重点实验室研究表明T7噬菌体颗粒很容易由免疫细胞吞噬并促进免疫细胞的成熟和NO等细胞因子分泌,可以作为细胞模型验证FMDV与免疫细胞的相互作用关系与作用强度。使用试剂盒检测培养液中的NO含量,可以反映出免疫细胞与抗原相互作用的强度。
[0381] TIM-3纳米抗体在高浓度时,对免疫细胞分泌NO的水平促进能力更好,解除了T细胞等免疫细胞受到的其他免疫细胞的抑制(通过TIM-3受体与配体的结合)。对免疫细胞的促进能力,表明了TIM-3纳米抗体有可以用于促进动物细胞免疫水平的提高,可以作为一种潜在的免疫佐剂或者有病毒传播时的免疫促进剂。TIM-3免疫抑制性受体也存在于其他免疫相关细胞中,阻断免疫检查点可以增强免疫相关细胞的活性已经验证清楚。
[0382] 2.3纳米抗体注射小鼠后,小鼠体内IL-4、IFN-γ细胞因子以及NO分泌变化水平
[0383] 使用索莱宝IL-4细胞因子检测试剂盒检测小鼠血清中IL-4细胞因子水平,索莱宝IFN-γ细胞因子检测试剂盒检测小鼠血清中IFN-γ细胞因子水平,索莱宝NO检测试剂盒检测小鼠血清中NO水平,结果见下表。(每组检测8只,每个数据代表了1只小鼠检测的OD450值。)
[0384] /CN111253489A/CN111253489A-39FM.gif)
[0385] 2.4金黄色葡萄球菌和无乳链球菌攻毒保护实验
[0386] 每组8只小鼠,攻毒24小时后小鼠状态,见下表。
[0387] /CN111253489A/CN111253489A-40FM.gif)
[0388] 对小鼠免疫水平的促进能力,揭示了TIM-3纳米抗体可以用于促进动物细胞免疫水平的提高,可以作为一种潜在的免疫佐剂或者有病毒/细菌传播时的免疫促进剂,用于动物的保护。TIM-3免疫抑制性受体也存在于其他免疫相关细胞中,虽然有很多机制没有研究清楚,但是阻断免疫检查点可以增强免疫相关细胞的活性已经验证清楚。本研究所制备的TIM-3纳米抗体可以用于保护动物在接受金黄色葡萄球菌和无乳链球菌侵害时,增强了小鼠存活的数量。
[0389] 以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精
神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。

